If you have or know someone with sickle cell disease, you know how debilitating it can be. Research into the condition has been going on for many years. Just recently, new data shows that a cure might be possible. Is this true? If so, what does it look like? How soon until it’s available to the general public?
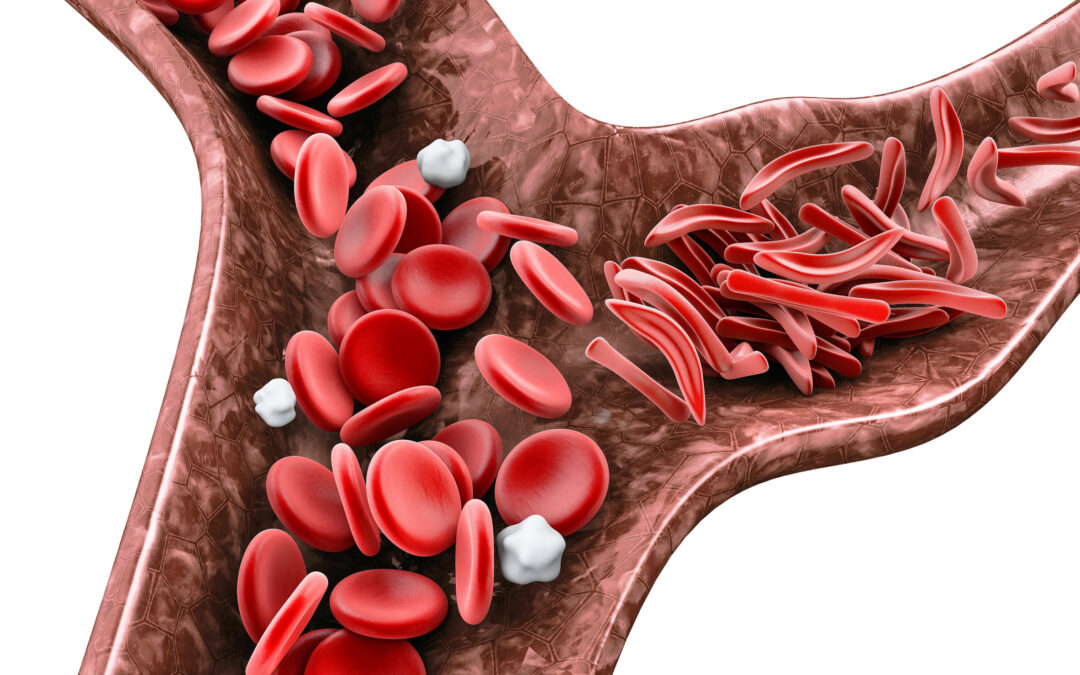
Sickle cell disease is a rare blood disorder caused by a genetic mutation that leads to the body making abnormal hemoglobin, the protein in red blood cells that carries oxygen around the body. Red blood cells are typically round and squishy, but mutated hemoglobin makes them rigid and take on a sickle, or crescent, shape.
As the sickled cells move throughout the body’s blood vessels, they get stuck, leading to blockages that stop the flow of blood. So, the risk of heart attack, stroke, and organ damage is much higher in people with sickle cell disease. Unfortunately, by age twenty or thirty, many patients have suffered strokes and been started on dialysis or have had their hips replaced and spleens removed. Individuals with severe cases have a life expectancy of about forty-five years.
A sickle-cell crisis is when a person experiences episodes of severe pain. It might be precipitated by infection, dehydration, stress, a change in seasons (winters were the worst since cold weather narrows the blood vessels near the skin and increases the risk of blockages), or nothing at all. Often a person in sickle cell crisis needs to be in the hospital to receive blood transfusions and high doses of opioid medications. Many people are in and out of the hospital every few months, some every couple of weeks.
The condition affects 100,000 Americans and millions of people globally, mainly in Africa. The Centers for Disease Control and Prevention (CDC) estimates that 1 in every 365 Black babies is born with the condition. Since it’s a genetic disorder, the disease runs in families.
Sickle Cell History
In September 1904, Walter Clement Noel, a twenty-year-old Grenadian man, started studying at the Chicago College of Dental Surgery. Even though few Black people were permitted to study at most American universities since Noel was well off, well educated, and a foreigner, he had secured a spot. During his journey from Barbados, he developed a painful sore on his ankle. He went to a doctor, who applied a tincture of iodine to the wound. The ulcer healed, leaving a scar like others on his body.
However, in November, he’d developed a cough and fever and had trouble breathing while he felt weak and dizzy. A few weeks later, he went to the hospital, where a medical resident named Ernest Irons studied his blood under a microscope. The findings were so unusual that Irons immediately alerted his supervising physician, James Herrick. Herrick later wrote that Noel’s blood had a “large number of thin, elongated, sickle-shaped and crescent-shaped forms.”
For the next two and a half years, Herrick and Irons followed Noel as he pursued his dental studies and suffered through joint problems, gastrointestinal upset, difficulty breathing, and episodes of severe pain. Despite their intense study, they failed to arrive at an explanation or a treatment for his condition. Noel finished school and returned to Grenada, where he practiced dentistry until he died of pneumonia at age thirty-two.
Other doctors read Herrick’s report and found patients of their own. By the 1920s, doctors determined sickle-cell disease to be a distinct, hereditary form of anemia. In 1949, the biochemist Linus Pauling published a paper linking the illness to hemoglobin. He described it as a “molecular disease.” However, molecular biology was in its infancy. In the decades following, sickle cell disease would be under-studied, largely because it primarily affects individuals of African descent.
If the sickling gene is so harmful, why do so many Black people carry it? In the early nineteen-fifties, a geneticist, Anthony C. Allison, found that the sickle-cell trait confers protection against malaria. When the malaria parasite infects the red blood cells of someone with the sickle cell trait, it has trouble hijacking the replication machinery. When looking at a map, the sickling gene appears to be most prevalent in regions where malaria is endemic, especially in southern Europe and Africa. Almost 80% of sickle-cell births occur in sub-Saharan Africa.
The Role of Hemoglobin
Hemoglobin is initially constructed using two alpha units and two gamma units. This is often referred to as fetal hemoglobin because, during the first few months of life, the body mostly stops producing gamma and starts making beta, which pairs with the alpha units forming adult hemoglobin. Fetal hemoglobin grabs hold of oxygen more tightly than adult hemoglobin, which is essential for a developing baby to take oxygen out of its mother’s bloodstream.
A single mutation on chromosome 11 (BCL11A) modifies the beta unit, resulting in hemoglobin S (a misshapen version that causes red blood cells to sickle). The mutation wasn’t identified until the mid-2000s. If a person inherits only one copy of this mutated gene, they have sickle cell trait and live relatively normal lives. Those who inherit two copies have sickle cell disease, and the effects can be devastating. Healthy red blood cells live for three or four months, whereas sickle cells die within weeks. Blood cells are made in our bone marrow, which works feverishly to create replacements but can’t keep up.
Treatment Options
Regarding treatments, only a handful of sickle cell medications have been approved by the Food and Drug Administration (FDA). For example, in 1998, the agency approved hydroxyurea, a cancer drug, to treat sickle cell disease. Somehow, the medication increases fetal hemoglobin production, decreasing the proportion of sickled hemoglobin in the bloodstream. Unfortunately, it has numerous side effects and doesn’t work for everyone.
Since 2017, three more medications have been approved for sickle cell disease: the amino acid L-glutamine, voxelotor (a hemoglobin-stabilizing drug), and crizanlizumab (a monoclonal antibody). While each has some benefits, they’re far from a cure. In addition, the latter two can cost a hundred thousand dollars a year.
For decades, bone marrow transplants have been a potentially curative option for patients with sickle cell. However, the process is risky and agonizing. The first step is to use high doses of chemotherapy to eliminate most of a patient’s existing bone-marrow cells. The second step is to transplant healthy donor stem cells into the bone marrow, where they can produce properly shaped hemoglobin.
To reduce the risk of transplant rejection, the donor is usually a family member, such as a sibling. However, less than 20% of sickle cell patients have a sibling who is a suitable match. So, many face some level of transplant rejection. All must take powerful immunosuppressive medications afterward, which places them at high risk of infection. As a result, only about 1200 procedures have been performed in the U.S. since 1984.
Advances in Gene Editing
In 2019, Dr. Haydar Frangoul, a pediatric hematologist at the Sarah Cannon Research Institute, tried a new transplant technique as part of a clinical trial. The process involved extracting the patient’s bone marrow cells from their blood, editing their DNA using the gene-editing technology CRISPR, and reintroducing the cells to the bone marrow. The cells had been modified so that they produced fetal hemoglobin. Since they were the patient’s own, there was little risk of rejection.
So far, the process has been tried on seven patients. After the therapy, all of the patients feel as though they’ve been cured because they haven’t had any sickle cell crisis episodes since the treatment. Researchers will follow these patients for 15 years to see if there are long-term complications and if they remain free of sickle cell and can be considered to be truly cured.
Another approach is to flush stem cells out of the bone marrow and into the bloodstream by giving the patient medication. Next, an apheresis machine collects the stem cells from the blood. A virus delivers microRNA (a short string of DNA) into cells from a patient’s bone marrow. The virus, or vector, permanently inserts the DNA into the cell’s genetic blueprint. The microRNA then interferes with the production of a protein that prevents fetal hemoglobin from being made, causing fetal hemoglobin production to turn back on.
A team at a Cambridge-based biotechnology company, Bluebird Bio, started a trial using this method in 2013. Since the trial began, it has treated dozens of patients; almost all have had their pain and other sickle cell complications eliminated.
One concern is that altering genes to rid them of sickle cell could introduce mutations that cause cancer at some point in the future. Bluebird Bio recently paused its clinical trial after two patients developed cancer. A further investigation cleared the gene therapy as the culprit, and the FDA allowed the studies to resume. Even before the cancer diagnoses, Bluebird revised its approach to improve efficacy. The company believes these changes have also made the therapy safer since no cancers have been identified in patients treated under the new processes.
Bluebird Bio expects to submit its gene therapy to the agency for approval next year. Also, Vertex Pharmaceuticals and CRISPR Therapeutics, the developers of the CRISPR technique, will ask that their treatment be reviewed shortly after that.
Some skeptics have pointed out that the cancer cases pose several vital questions. Are some people with sickle cell disease at increased risk for leukemia? Is their bone marrow potentially unsuitable for these types of interventions? It’s not new that chemotherapy used for transplants can cause secondary cancers. So, the risk could be considerably higher for sickle-cell patients whose bone marrow already contends with high levels of stress. This is one of the reasons why many scientists caution that it’s too early to say that gene therapy is a cure, but the early signs are encouraging.
Factors Hindering the Progress
A major hurdle for gene therapy is the expected price tag of $1 million to $2 million per patient. However, compared to data from private insurers that show the lifetime cost for a person with sickle cell is $2.7 million, the gene therapy price doesn’t seem as expensive.
The National Institute of Health (NIH) has created two independent committees to study how much money could be saved over a lifetime of treatment for sickle cell if people with the disease could be cured using gene therapy. Some experts note the price of a potentially curative one-time treatment would be significantly less than the lifetime cost of caring for people with the disease.
Officials at the Centers for Medicaid and Medicare Services (CMS) are looking at the possibility of covering the cost of experimental therapies for patients. The federal government has paid such costs before for bone-marrow transplants for sickle cell, and amyloid PET brain scans for Alzheimer’s disease. Currently, Medicaid programs cover about half of sickle cell patients in the U.S. However, the programs differ state by state, and some states with relatively large Black populations have among the most constrained budgets.
Money isn’t the only obstacle. In many parts of the country, especially poor or rural areas, there’s a lack of hematology expertise. Also, many sickle cell patients lack access to regular primary care, so they’re forced to rely on emergency departments when serious problems arise.
Another issue is distrust. For good historical reasons, many minority communities have a deep suspicion of research, scientists, and doctors. There’s an even greater suspicion of novel experimental treatments. Roughly 7 in 10 Black Americans think the health system mistreats people based on race. This number isn’t surprising when you consider Black women are over three times as likely as white women to die during childbirth, and Black men live a half-decade less than white men on average. During the Covid-19 pandemic, Black Americans were killed at twice the rate of white Americans.
While new gene therapies hold extraordinary promise, they must overcome a medical system that marginalizes Black Americans to be able to help those that need it the most.
The supreme goal is to find a simple, cheap, and effective way to cure sickle cell with a gene therapy that could be injected like a vaccine. This would allow it to be used in developing countries where complex, costly gene therapies would be unaffordable or impractical. To reach that target, the NIH is working with the Gates Foundation, academic researchers, several small biotechs, and the giant drug company Novartis. However, it may be a decade before this happens.
When it comes to curing sickle cell disease, we’re fortunate that it’s caused by a single gene mutation, resulting in a single change in a single protein. Some other genetic diseases cause dozens of mutations on separate genes or chromosomes, increasing the complexity of correction. It’s also opportune that it’s a disorder of the blood and bone marrow because, compared with other organs, it can more easily be extracted, modified, and replaced.
The clinical trials of gene therapies for sickle cell have produced astonishing results, raising hopes that a cure could be at hand for the many people with the disease. While the treatments won’t be cheap, and many people who need them the most are on the fringes of the medical system, if our experience with Covid-19 vaccines has taught us anything, it’s that creating a medical innovation and reaching those who need it are separate missions. However, with the proper funding and support, it is possible!